Infection with the carcinogenic liver fluke Opisthorchis viverrini modifies intestinal and biliary microbiome.
Jordan L. Plieskatt, Raksawan Deenonpoe, Jason P. Mulvenna, Lutz Krause, Banchob Sripa, Jeffrey M. Bethony and Paul J. Brindley. (2013) The FASEB Journal, vol. 27 no. 11 4572-4584.
Project Summary
Opisthorchis viverrini is a fish-borne trematode endemic in East Asia. Following ingestion, the flukes locate the biliary tree where chronic infection frequently leads to cholangiocarcinoma (CCA). The mechanisms by which O. viverrini infection culminates in CCA remain unknown. An unexplored aspect is its influence on the host microbiome. In the hamster, infection with this pathogen reliably leads to CCA.
In this study, genomic DNAs of microbiota from colorectal contents and bile of hamsters and from whole O. viverrini were examined in this model of fluke-induced CCA. Microbial communities were characterized by high-throughput sequencing of variable regions 7-9 of prokaryotic 16S ribosomal DNA. Of ~1 million sequences, 536,009 with useable reads were assignable to 29,776 operational taxonomy units (OTUs) and, in turn, to 20 phyla and 273 genera of Bacteria or Archaea. Microbial community analyses revealed that fluke infection perturbed the gastrointestinal tract microbiome, increasing Lachnospiraceae, Ruminococcaceae, and Lactobacillaceae, while decreasing Porphyromonadaceae, Erysipelotrichaceae, and Eubacteriaceae (P <= 0.05). More than 60 OTUs were detected in the biliary system, which confirmed bacteriobilia and a noteworthy community of microbes associated with the parasites. The fluke-associated microorganisms included potential pathogens from the Enterobacteriaceae and Listeriaceae and others, including Cyanobacteria and Deinococci, usually found in external environments. Given that opisthorchiasis is distinguished from other helminth infections by a robust inflammatory phenotype with conspicuously elevated IL-6, and that inflammation of the biliary system leads to periductal fibrosis, which is a precursor of CCA, the flukes and their microbiota may together drive this distinctive immune response.
Resources
Full manuscript is available at http://www.fasebj.org/content/27/11/4572.long
Unprocessed datasets can be downloaded from the SRA at http://www.ncbi.nlm.nih.gov/sra/?term=SRA065893
Selected results from the manuscript
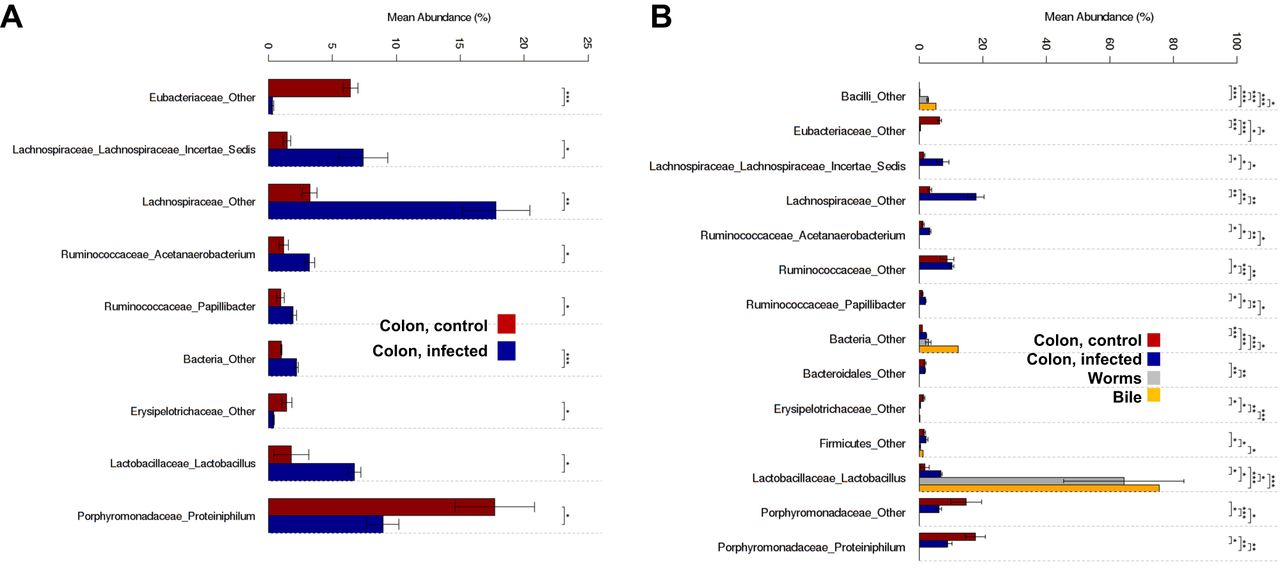
Figure 3: Significant differences were evident among the microbial communities in the control hamsters and in the hamsters infected with O. viverrini. Community compositions are presented for families/genera of microorganisms that exhibited differences among groups. A) Bacterial composition (mean abundance) of colorectal feces in the control and O. viverrini-infected hamsters. B) Bacterial composition of the colorectal contents (feces) of the control and O. viverrini-infected hamsters, of the whole O. viverrini worms, and of the bile from infected hamsters. *P <= 0.05, **P <= 0.01, ***P <= 0.001.
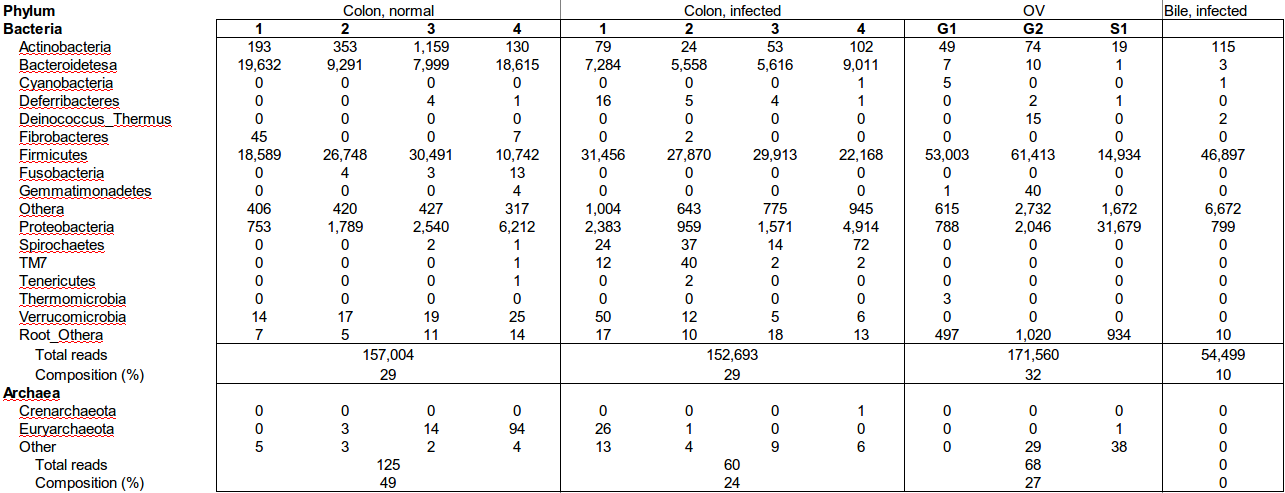
Table 1: Bacteria and Archaea phyla and number of reads detected in hamster colon contents, hamster bile, and whole O. viverrini worms by pyrosequencing of amplicons targeting prokaryotic 16S rRNA genes.
Supporting Table S1A: OTUs where numbers were much higher then the biliary system (in worms and bile) in comparison to colorectal feces of hamsters (infected and control). Members of the order Lactobacillales were the most abundant OTUs in the worms and bile.
Actinomycetales_Propionibacterineae
Archaea_Other
Bacillales_Other
Bacilli_Other
Bacteria_Other
Burkholderiaceae_Cupriavidus
Clostridiaceae_"Clostridiaceae 1"
Comamonadaceae_Comamonas
Comamonadaceae_Hydrogenophaga
Comamonadaceae_Other
Enterobacteriaceae_Citrobacter
Enterobacteriaceae_Other
Gammaproteobacteria_Other
Gemmatimonadaceae_Gemmatimonas
Incertae sedis 5_Other
Incertae sedis 5_Tepidimonas
Lactobacillaceae_Lactobacillus
Lactobacillales_Other
Moraxellaceae_Acinetobacter
Moraxellaceae_Alkanindiges
Planococcaceae_Jeotgalibacillus
Planococcaceae_Other
Pseudomonadaceae_Chryseomonas
Pseudomonadaceae_Flavimonas
Pseudomonadaceae_Pseudomonas
Root_Other
Sphingomonadaceae_Sphingomonas
Supporting Table S1B: OTUs detected by pyrosequencing using broad range 16S rRNA gene primers in the biliary system - in O. viverrinidd worms and bile - but not in the colorectal feces of the hamsters (infected or control). Not only were these OTUs from numerous phyla of the Bacteria and Archaea domains, they also included phylotypes from genera that are potential pathogens in humans, including Bordetella, Brochothrix, Burkholderia, Leminorella, Pseudomonas, Serratia, and Sphingomonas, all of which are gram-negative or -positive bacteria. This biliary tract microbiota also included microbes known usually from the external environment, including freshwater, soil, and even volcanic springs: Cyanobacteria, Methylobacterium, Mesorhizobium, Flavobacterium, Truepera, and others.
Acetobacteraceae_Muricoccus
Acetobacteraceae_Roseomonas
Aeromonadales_Other
Alcaligenaceae_Bordetella
Alcaligenaceae_Pusillimonas
Anaplasmataceae_Neorickettsia
Bacillaceae_"Bacillaceae 1"
Bacillariophyta_Other
Bradyrhizobiaceae_Bradyrhizobium
Burkholderiaceae_Burkholderia
Burkholderiaceae_Other
Burkholderiaceae_Ralstonia
Caulobacteraceae_Brevundimonas
Caulobacteraceae_Other
Caulobacteraceae_Phenylobacterium
Chromatiaceae_Rheinheimera
Comamonadaceae_Caldimonas
Comamonadaceae_Diaphorobacter
Comamonadaceae_Variovorax
Coxiellaceae_Aquicella
Crenotrichaceae_Terrimonas
Deinococcaceae_Deinococcus
Enterobacteriaceae_Leminorella
Enterobacteriaceae_Rahnella
Enterobacteriaceae_Raoultella
Enterobacteriaceae_Salmonella
Enterobacteriaceae_Serratia
Euryarchaeota_Other
Flavobacteriaceae_Cloacibacterium
Flavobacteriaceae_Flavobacterium
Flexibacteraceae_Niastella
GpIIa_Other
Halomonadaceae_Halomonas
Hyphomicrobiaceae_Devosia
Incertae Sedis XII_Exiguobacterium
Incertae Sedis XI_Anaerococcus
Incertae Sedis XVII_Thermaerobacter
Incertae sedis 5_Leptothrix
Listeriaceae_Brochothrix
Methylobacteriaceae_Methylobacterium
Moraxellaceae_Other
Moraxellaceae_Psychrobacter
Oceanospirillaceae_Other
Pasteurellaceae_Lonepinella
Phyllobacteriaceae_Mesorhizobium
Phyllobacteriaceae_Other
Planococcaceae_Planomicrobium
Porphyromonadaceae_Porphyromonas
Pseudomonadaceae_Cellvibrio
Pseudomonadales_Other
Rhizobiaceae_Agrobacterium
Rhodobacteraceae_Other
Rhodobacteraceae_Paracoccus
Rhodobacteraceae_Rubellimicrobium
Rubrobacterales_Rubrobacterineae
Sphingomonadaceae_Novosphingobium
Sphingomonadaceae_Sphingobium
Trueperaceae_Truepera
Xanthomonadaceae_Other
Xanthomonadaceae_Pseudoxanthomonas
Xanthomonadaceae_Stenotrophomonas
Xanthomonadaceae_Thermomonas

